Fenilchetonuria (PKU)
Che cos’è la fenilchetonuria?
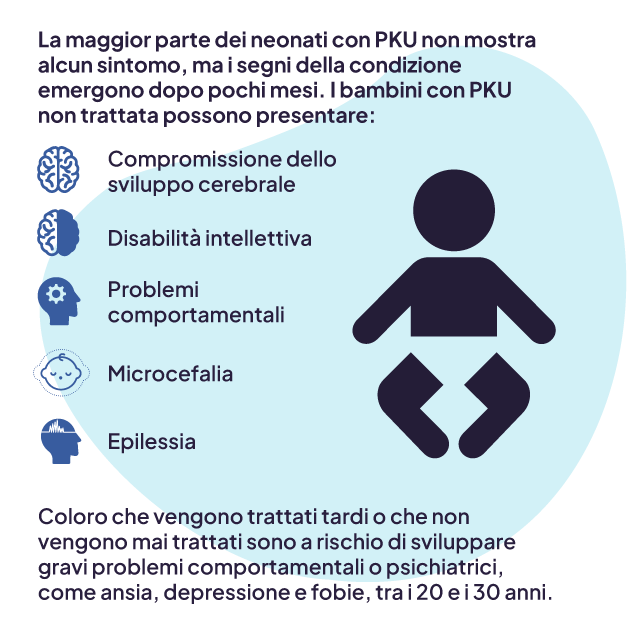
La fenilchetonuria (PKU) è una rara malattia metabolica ereditaria che colpisce il cervello.1 È causata da un difetto nel gene che aiuta a produrre l’enzima necessario per degradare la fenilalanina.1 Se non trattata o gestita in modo superficiale, la fenilalanina, un aminoacido essenziale presente in tutte le proteine e nella maggior parte degli alimenti, può accumularsi fino a raggiungere livelli nocivi per l’organismo. Ciò può causare disabilità gravi e irreversibili, come disabilità intellettiva permanente, crisi convulsive, ritardo dello sviluppo, perdita della memoria e problemi comportamentali ed emotivi.1
I neonati con fenilchetonuria non presentano sintomi inizialmente, ma questi sono generalmente progressivi e i danni causati dai livelli tossici di fenilalanina nei primi anni di vita sono irreversibili.2,3
La fenilchetonuria viene diagnosticata solitamente attraverso il programma di screening neonatale.4
Quanto è comune la fenilchetonuria?
Si stima che vi siano 58.000 persone affette da fenilchetonuria a livello globale. Maschi e femmine hanno la stessa probabilità di ereditare il gene difettoso e di sviluppare la fenilchetonuria.


Neil Smith Global PKU Project Leader in PTCSappiamo, grazie alle nostre ricerche, che esiste una vasta comunità di pazienti affetti da PKU che necessita di ulteriori opzioni terapeutiche. Abbiamo inoltre instaurato rapporti straordinari all'interno di questa comunità, sia con i pazienti stessi che con le organizzazioni di supporto. Aiutare questa comunità è la mia missione e la mia motivazione, la mia fiducia nella scienza mi aiuta a rimanere concentrato e mi spinge ad andare avanti.
[1] de Groot MJ, Hoeksma M, Blau N, et al. Mol Genet Metab 2010;99:S86–S89.
[2] Phenylketonuria (PKU). Disponibile all’indirizzo: https://www.mayoclinic.org/diseases-conditions/phenylketonuria/symptoms-causes/syc-20376302. Ultimo accesso: ottobre 2021.
[3] Blau N, van Spronsen FJ, Levy HL. Lancet 2010;376:1417–1427.
[4] Al Hafid N, Christodoulou J. Transl Pediatr 2015;4(4):304–317.